[논문] The shaky foundations of simulating single-cell RNA sequencing data(2)
The shaky foundations of simulating single-cell RNA sequencing data
[논문] The shaky foundations of simulating single-cell RNA sequencing data(2)
The shaky foundations of simulating single-cell RNA sequencing data
Discussion
- 시뮬레이터 평가로 gene/cell/global summary 기준으로 비교, 전체 성능 순위화
- ZINB-WaVE는 거의 모든 평가에서 좋은 성능
- 시뮬레이터와 벤치마킹의 신뢰성 문제
- 벤치마크 평가를 위해 ground trurh가 필요한데, 실험적으로 정답을 확보하기 어려운 경우를 위해 유연하고 신뢰성 있는 시뮬레이션 프레임워크 개발 필수적
- 시뮬레이터를 사용한 분석의 한계
- 대부분 시뮬레이터는 단일 그룹(한 개의 클러스터/배치)만 생성 가능.
- 실제 데이터와 비교 시 클러스터링 결과 왜곡, summary redundancy 존재
- 품질 평가 도구(scater, countsimQC) 활용의 부족. 시뮬레이터를 개발하거나 사용할 때 참조 데이터와 비교한 요약 리포트가 함께 제공되면 신뢰도 향상
- 클러스터링의 경우 시뮬레이션 기반 평가에서는 과대평가
- 성능 외 고려할 요소
- splatter는 사용하기 쉽고 문서화가 잘 되어있으나 다른 시뮬레이터는 성능은 좋더라도 사용성이 낮고 파라미터 해석이 어려움
- 목적별로 중요한 summary 지표를 정의하고, 평가 리포트를 표준화할 필요성
- trajectory 기반 시뮬레이터도 존재하기에 trajectory 관련 신뢰성 확보 필요(해당 연구는 type n, b, k로 시뮬레이터들을 분류)
- 향후 유연하고 해석 가능한 시뮬레이터 필요
Conclusions
- 현재 시뮬레이터들의 한계
- 표현할 수 있는 데이터 복잡성 수준에 제한이 있음
- 유전자 발현 차이를 만들기 위해 사용자의 인위적 입력에 의존함
- 시뮬레이터가 얼마나 현실적인 데이터 특성을 반영할 수 있느냐에 따라, 다른 분석 도구들을 평가하는 데 적합한 정도도 달라짐
- 시뮬레이션 기반 벤치마크 연구 결과는 사용하는 시뮬레이터에 따라 달라지며 시뮬레이터의 성능이 높다고 해서 integration이나 클러스터링 방법의 평가 결과의 신뢰도가 높지 않음
- 어떤 품질 지표(summary)를 어떻게 선택하느냐가 평가 결과에 영향을 미침
- scRNA-seq 데이터 구조를 충실하게 반영할 수 있는 summary의 종류, 개수, 중요도를 정의하는 연구 필요
Methods
Reference datasets
- 데이터의 출처는 GitHub, GEO(혹은 GSE, Gene Expression Omnibus), Biocunductor의 ExperimentHub 등
raw 데이터는 R에서 사용되는 SingleCellExperiment 클래스 형식으로 변환, 그 외는 대부분 전처리 없이 사용
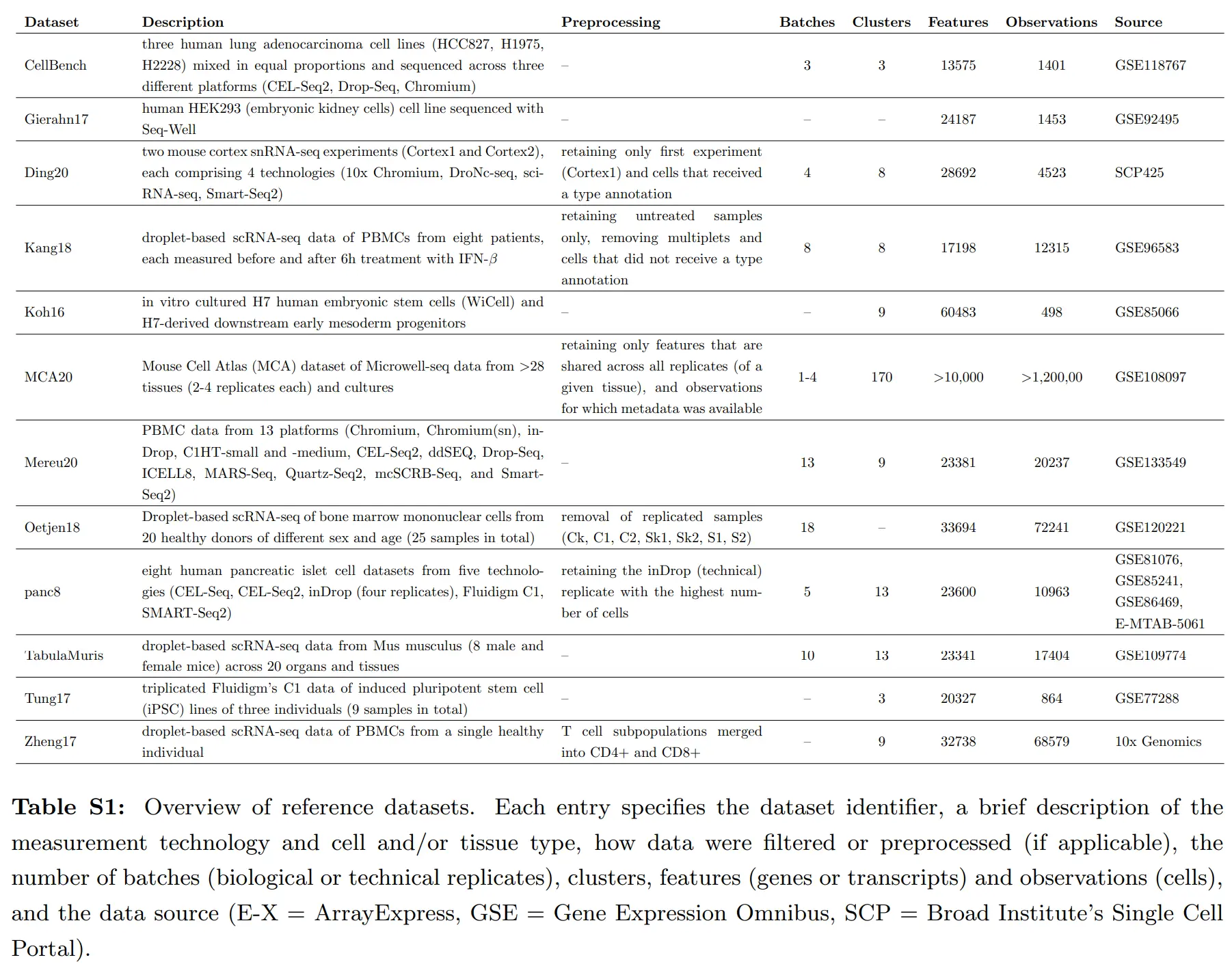
- 참조 데이터는 최소한의 필터링만 수행
- 세포 수가 너무 적은 클러스터/배치 제거 및 감지율이 낮거나 total count가 적은 유전자/세포 제거
- 다양한 부분 집합(subset) 샘플링해 배치/클러스터 수 제어
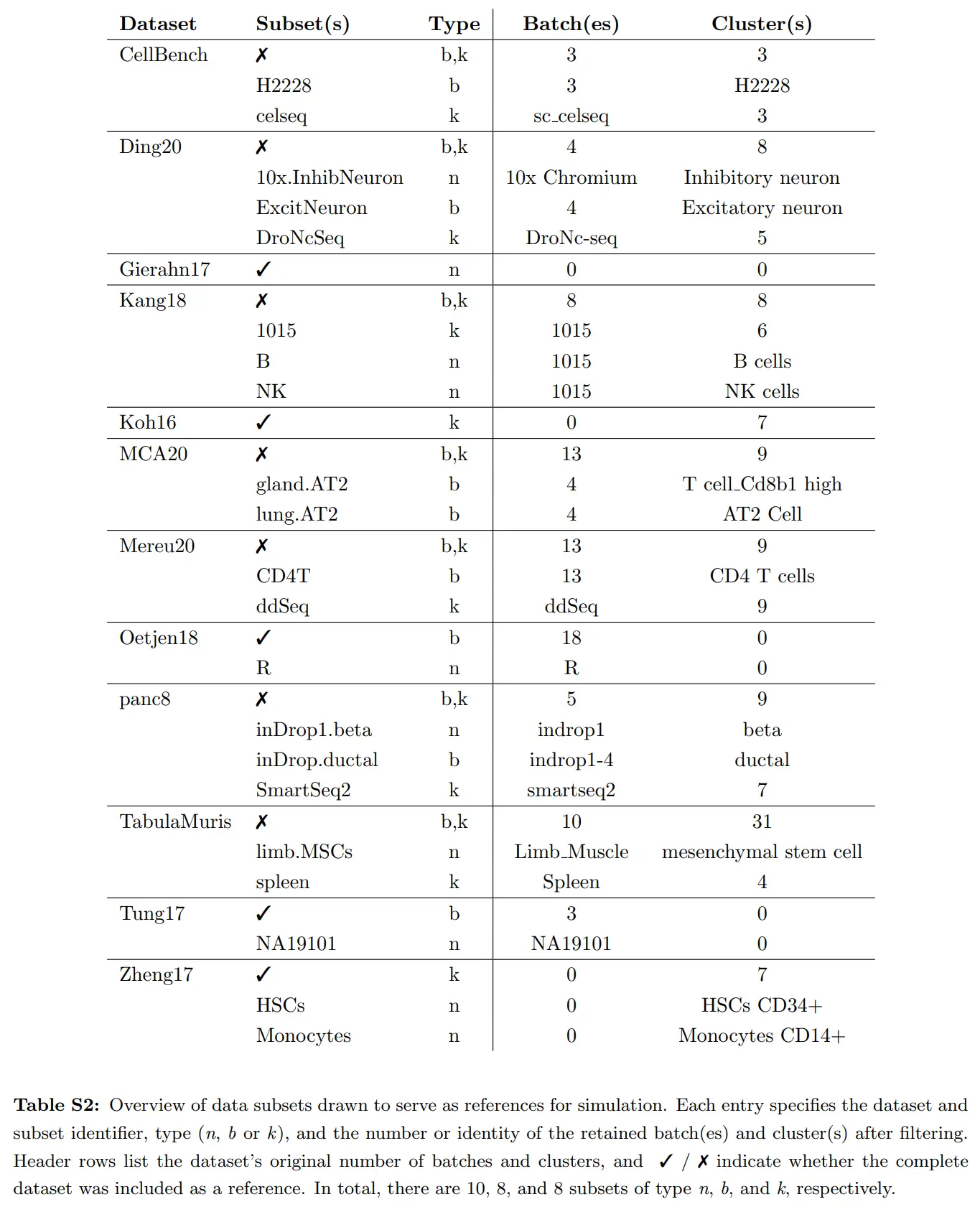
Simulation methods
- 대부분 기본 설정값을 사용. 저자가 제시한 권장사항이 있다면 그에 따라 설정
각 시뮬레이션 방법의 모델 구조와 병렬처리 지원 여부에 대한 간략한 개요
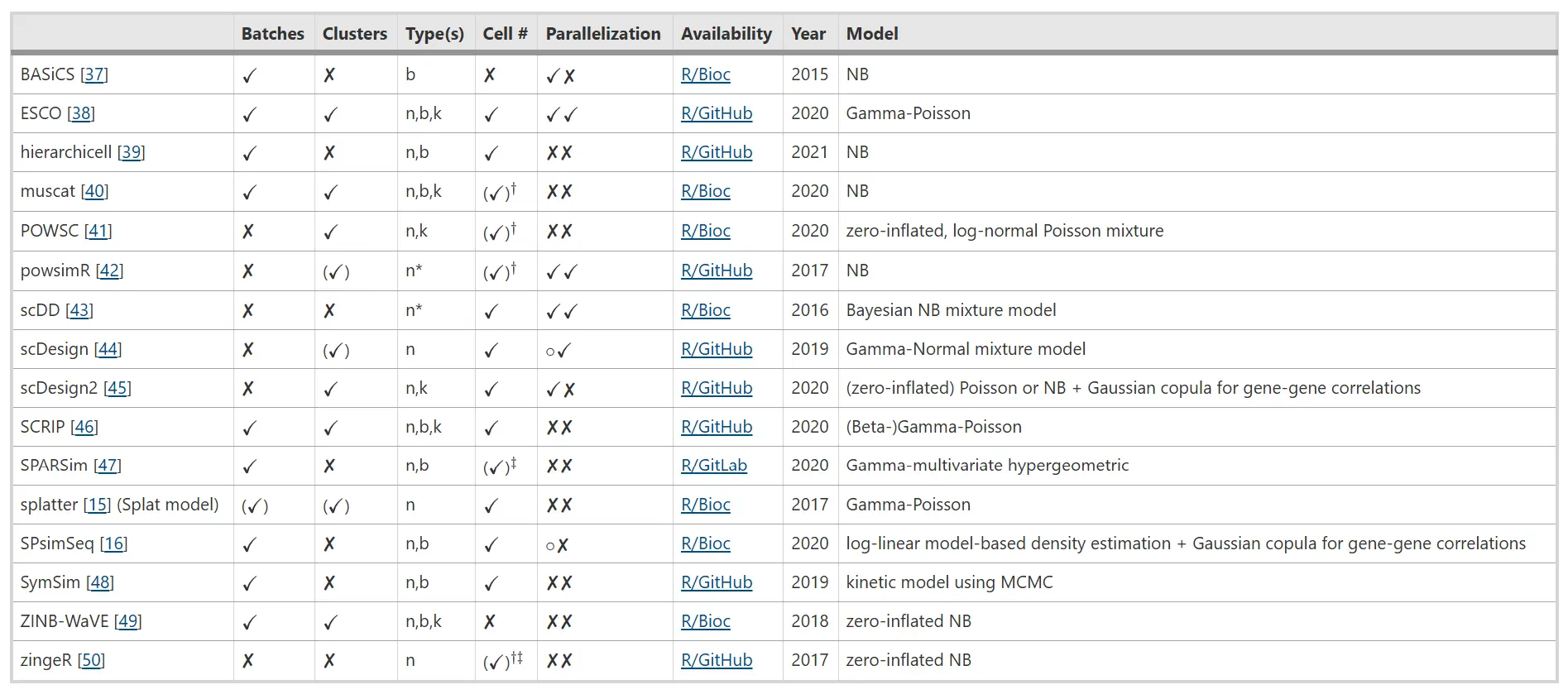
- 파라미터 추정과 시뮬레이션 실행에 사용된 실제 인자
Quality control summaries

- gene-level의 5개 summary 지표 계산
- logCPM의 average
- logCPM:
scater패키지의calculateCPM함수로 계산한 CPM(counts per million)을 log1p(log(1+x)) 변환한 값
- logCPM:
- logCPM의 variance
- CV(coefficient of variation): 변이 계수
- detection frequency: 0이 아닌 값이 있는 세포의 비율
- gene-to-gene correlation
- logCPM의 average
- cell-level의 6개 summary 지표 계산
- library size: 세포별 총 카운트 (read 수)
- detection frequency: 해당 세포에서 발현된 유전자의 비율
- cell-to-cell correlation: logCPM 기반 세포 간 유사도
- cell-to-cell distance: PCA 공간에서 유클리디안 거리
- KNN occurrences: 해당 세포가 다른 세포의 K-최근접 이웃으로 몇 번 등장했는지
- local density factors (LDF): PCA 공간에서 세포의 주변 밀도를 측정한 상대값 (그룹 구조 파악용)
- LDF: 그룹 구조(batch 또는 cluster)를 수치적으로 평가하는 데 목적
- PCA 기반 summary 계산 절차
scran패키지의moleGeneVar를 logCPM에 적용- 변이가 큰 유전자 500개(HVGs) 선택(
getTopHVGs) scater의calculatePCA로 첫 50개의 주성분(PCs) 계산
- type n 이외의 데이터에서는 다음 기준으로 계산
- global
- batch-level
- cluster-level
- global의 3개 summary 지표 계산
- percent variance explained (PVE)
- PVE: 발현량의 전체 분산에서, 세포가 속한 그룹(배치/클러스터)로 설명되는 비율
- cell-specific mixing score (CMS): 세포가 다른 그룹과 얼마나 섞여 있는지 측정
- silhouette width: 클러스터의 경계 명확도 (내부 vs 외부 거리 차이)
- percent variance explained (PVE)
Evaluation statistics
- 각 summary 지표에 대해 참조 데이터와 시뮬레이션 데이터 간의 차이 평가 방법
- Kolmogorov-Smornov(KS) 통계량:
stats패키지의ks.test()함수 사용 - Wasserstein 거리(Earth Mover’s Distance (EMD)):
waddR의wasserstein_metirc()함수 사용
- Kolmogorov-Smornov(KS) 통계량:
- 2차원 지표 간의 결합 분포를 비교
- 2차원 KS 통계량:
MASS패키지의kde2d()함수 사용 - 2차원 EMD:
emdist패키지의emd2d()함수 사용 - 2차원 비교는 유전자 수준과 세포 수준 summary 조합 중 의미 있는 것만 사용하며, Global summary와 상관계수 기반 summary는 제외
- 2차원 KS 통계량:
Runtime evaluation
- 시뮬레이터의 실행 시간 정량화
- 각 시뮬레이션 방법과 서브셋 조합에 대해 측정
- 파라미터 추정 시간
- 데이터 생성(시뮬레이션) 시간
- 각 시뮬레이션 방법과 서브셋 조합에 대해 측정
Integration evaluation
- batch 통합 방법의 평가 방식
- 데이터 통합(Data Integration): 서로 다른 조건/배치/플랫폼에서 얻은 단일세포 데이터를 하나의 통합된 공간에서 비교/분석 가능하도록 정렬하는 과정
- 공간 변환 기반: 차원 축소된 표현 공간에서 서로 다른 배치 데이터를 정렬(PCA, Harmony, fastMNN, CCA 등)
- 표현학습 기반: VAE, autoencoder 등 딥러닝 기반 표현 학습(scVI, totalVI 등)
- 유전자 수준 정규화: Raw count에서 배치별로 보정(ComBat, limma)
- 사용된 통합 분석 방법(additional file1의 sec 5.1 참고)
- ComBat
- Harmony
- fastMNN, mnnCorrect
- limma
- Seurat
| 도구/방법 | 설명 | 특징/장점 | 주요 사용 환경 |
|---|---|---|---|
| ComBat | Empirical Bayes 기반의 배치 보정 방법. 원래는 microarray/bulk RNA-seq용으로 개발됨. | 빠르고 간단, 연속형 및 이산형 배치 변수 처리 가능 | Bulk RNA-seq, scRNA-seq의 pseudobulk에도 사용 가능 |
| Harmony | Embedding 공간(예: PCA)에서 batch effect를 제거하는 알고리즘. 반복적 군집화 기반. | 빠르고 scalable. 클러스터 구조를 보존하면서 batch 제거 | Seurat, Scanpy 등과 함께 사용됨 |
| fastMNN / mnnCorrect | Mutual Nearest Neighbors(MNN)를 이용한 배치 정렬 방법. fastMNN은 mnnCorrect의 고속 버전. | 비선형 구조 잘 보존, 여러 배치 간 연속적인 통합 가능 | scran, batchelor 패키지에서 사용 (R 기반) |
| limma | 선형 모델 기반의 분석 패키지. batch 효과도 모델링 가능함. | 통계적 안정성 높고 다양한 공변량 함께 처리 가능 | Bulk RNA-seq 중심이지만, scRNA-seq pseudobulk에 사용 |
| Seurat (Integration) | Canonical Correlation Analysis (CCA), RPCA 등으로 다양한 방식의 데이터 통합 지원 | 사용 편의성 높음, 시각화와 통합 분석까지 일괄 처리 가능 | R 기반 single-cell 분석의 대표 툴 |
- 방법의 성능 평가
- CMS(Cell-specific Mixing Score):
CellMixS패키지의cms()함수 계산- CMS ⟶ CMS*: 0.5를 빼서 평균이 0이 되도록 조정
- LDF 차이(Local Density Factor Difference):
ldfDiff()함수 사용- LDF ⟶ LDF*: 평균 0, 값의 범위 0 ~ 1로 스케일 조정
- ⟶ 서로 다른 세포들이 서로 다른 배치 간에 얼마나 잘 섞였는지 평가하는 지표. 값이 0에 가까울수록 잘 섞인 상태
- 최종 통합 점수(Overall intergration score)는 CMS*와 LDF*를 단순 평균한 값으로 계산
- CMS(Cell-specific Mixing Score):
- 각 배치 내에서 세포 단위 평균 계산, 이후 배치 간 평균 계산
Clustering evaluation
- 클러스터링(세포 분류) 방법(additional file1의 sec 5.2 참고)
- CIDR
- 계층적 클러스터링(HC)
- 주성분 분석(PCA)을 이용한 k-평균 클러스터링(KM)
- pcaReduce
- SC3
- Seurat
- TSCAN
- t-SNE 기반의 KM 클러스터링
| 방법 | 설명 | 특징/장점 | 주요 알고리즘/기술 |
|---|---|---|---|
| CIDR | Dropout을 고려하여 PCA 기반 거리 계산 후 클러스터링 수행 | Dropout 보정 내장, 빠르고 정확도 높음 | PCA + hierarchical clustering |
| 계층적 클러스터링 (HC) | 데이터 간 유사도 기반으로 트리 형태로 군집화 | 시각화 직관적, 군집 수 지정 없이 시작 가능 | 거리 행렬 + linkage method |
| PCA + K-평균 (KM) | PCA로 차원 축소 후 K-means로 클러스터링 수행 | 계산 빠름, 시각화와 연계 쉬움 | PCA + K-means |
| pcaReduce | PCA를 반복적으로 수행하면서 클러스터 개수를 줄여가는 방식 | 군집 수를 자동으로 줄여가며 구조 탐색 | Hierarchical PCA-based reduction |
| SC3 | 여러 클러스터링 결과를 통합하는 consensus clustering 기법 | 안정적이고 정확도 높음, 시각화 지원 | Multiple distance metrics + consensus |
| Seurat | 그래프 기반 Louvain/Leiden 알고리즘을 이용한 클러스터링 | 매우 널리 사용됨, 통합 분석까지 가능 | SNN graph + Louvain/Leiden |
| TSCAN | pseudotime 추정과 클러스터링을 결합한 분석 기법 | 궤적 기반 클러스터링, 시간 흐름 해석 가능 | MST (Minimum Spanning Tree) |
| t-SNE + K-평균 | t-SNE로 시각화한 저차원 공간에서 K-means 클러스터링 수행 | 시각화 직관적, 비선형 구조 반영 가능 | t-SNE + K-means |
- 적용 가능한 경우 클러스터의 개수는 실제 클러스터 수와 일치하도록 설정
- 방법의 성능 평가
- Hungarian 알고리즘(예측된 라벨과 실제 라벨 간 최적 매칭을 찾아서 정확도를 계산): 실제 클러스터 라벨과 예측된 클러스터 라벨 매칭
- 클러스터 단위의 정밀도(precision), 재현율(recall), F1 score 계산
Availability of data and materials
- Raw data
- 10x Genomics
- ArrayExpress
- Broad Institute의 Single Cell Portal(SCP)
- Gene Expression Omnibus(GEO)
- Zenodo의 R 객체(.rds 파일): 연구 주요 결과 재현(additional file1의 sec 7 참고)
- 참조 및 시뮬레이션 데이터에 대한 전반적인 품질 관리 요약(glbal and gene- and cell-level QC)
- 모든 데이터셋과 방법에 대해 계산된 1차 및 2차원 테스트 통계량
- 클러스터링 및 통합 결과
- 각 데이터 유형에 대해 1개 데이터셋에 대한 유전자 및 세포 subset별 5회 반복 수행한 runtimes
- 분석 환경: R v4.1.0 및 Bioconductor v3.13
- 계산 워크플로우: Snakemake v5.5.0, Python v.3.6.8
presentation
This post is licensed under CC BY 4.0 by the author.